 2025 Newsletter
2025 Newsletter
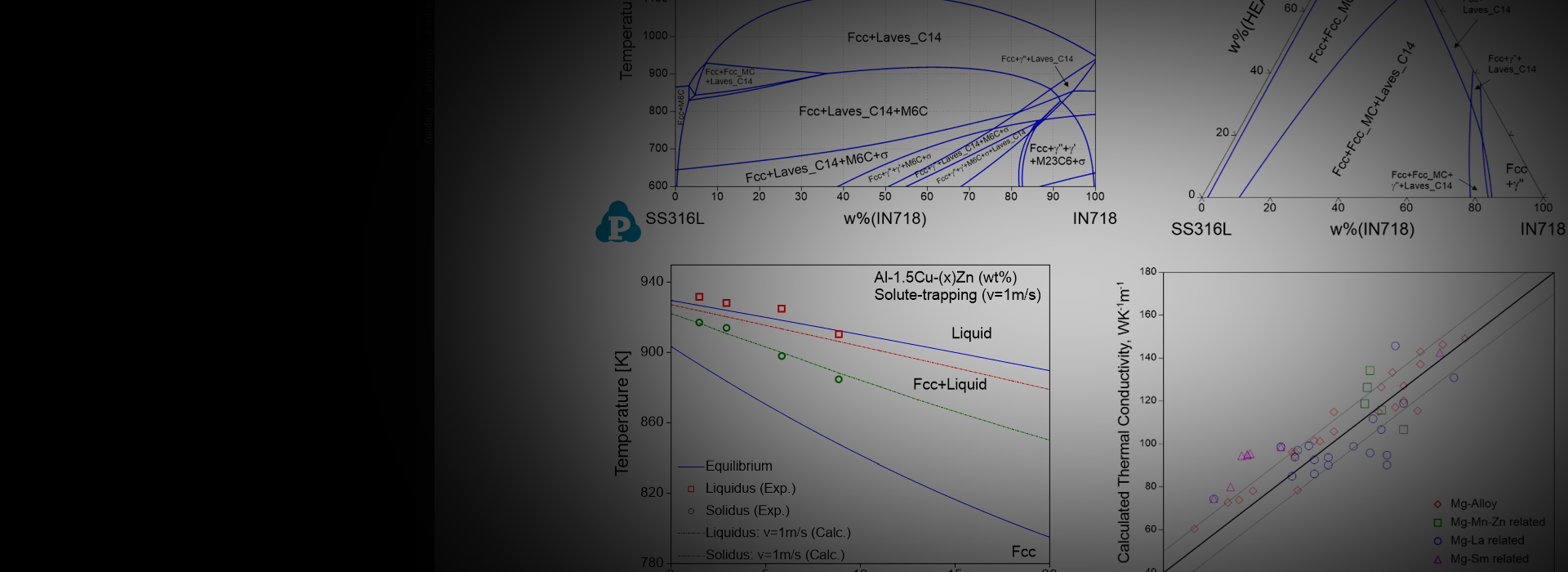
Material to Material Calculations
Solute Trapping Model for PanSolidification Simulations
New Databases for Molten salt and Thermal Conductivity
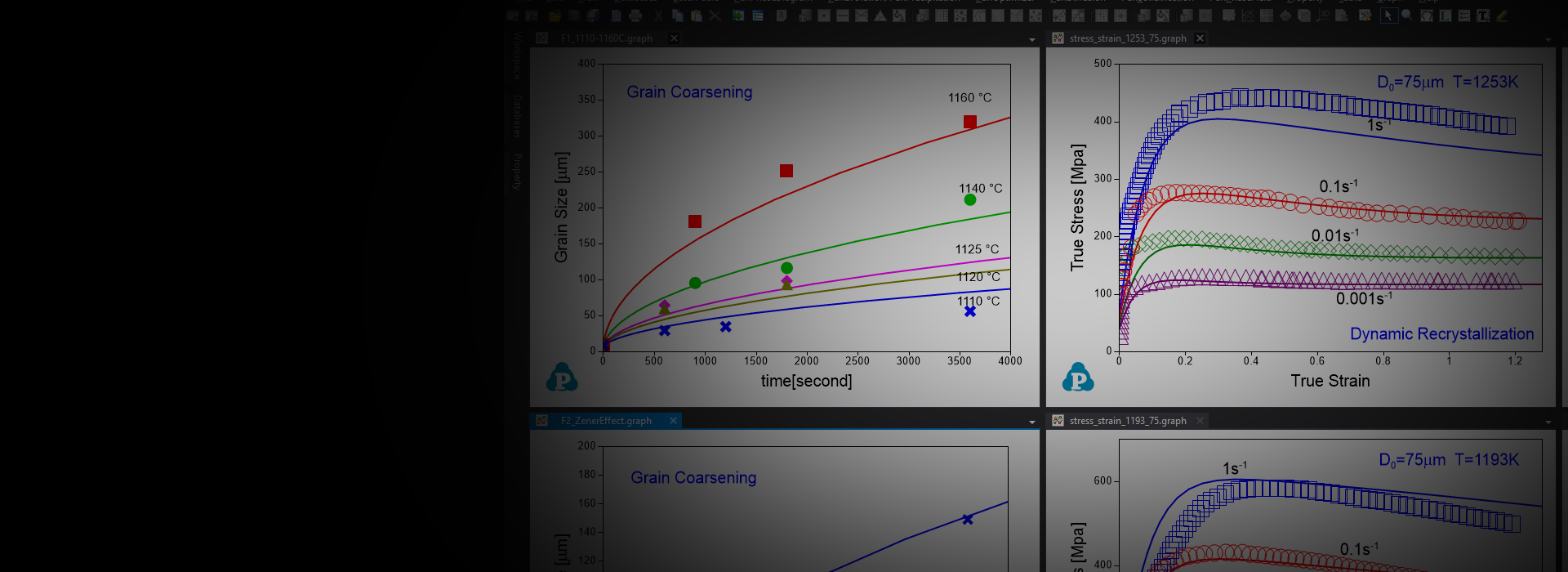
Concurrent Grain Growth and Precipitation Simulation
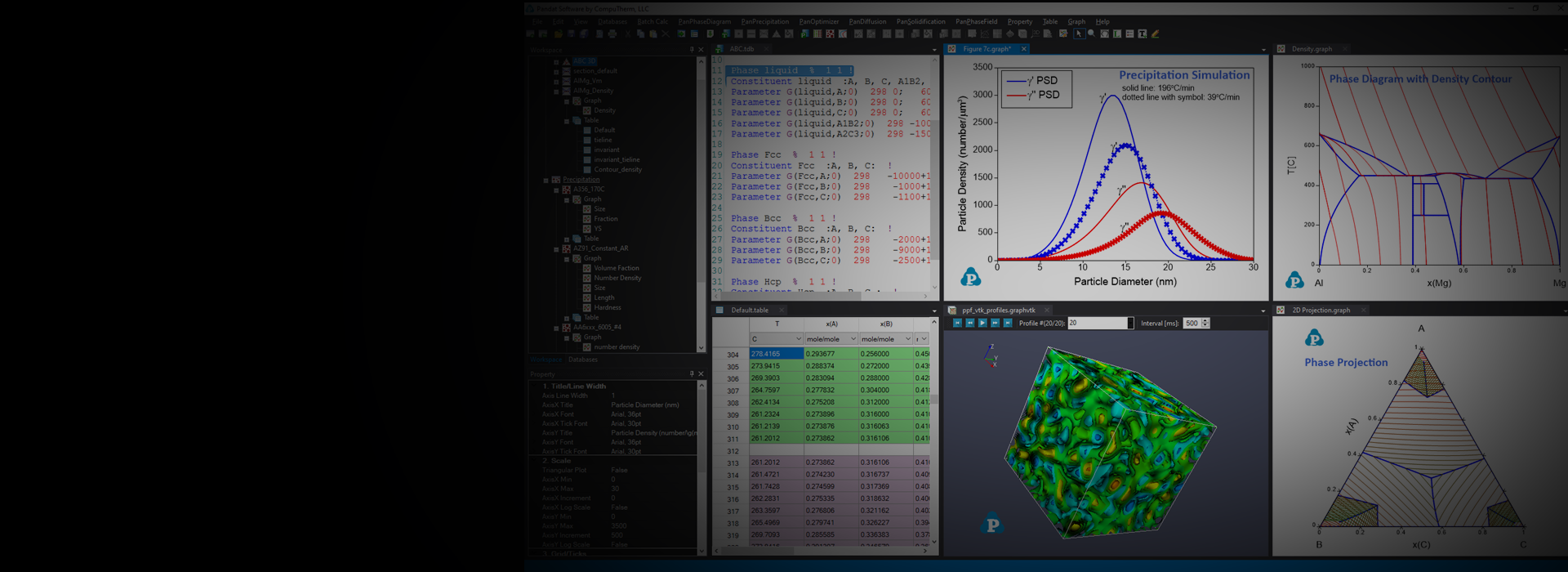
Precipitation Simultion
TTT and CCT Curves
Easy Coupling with Other ICME Tools
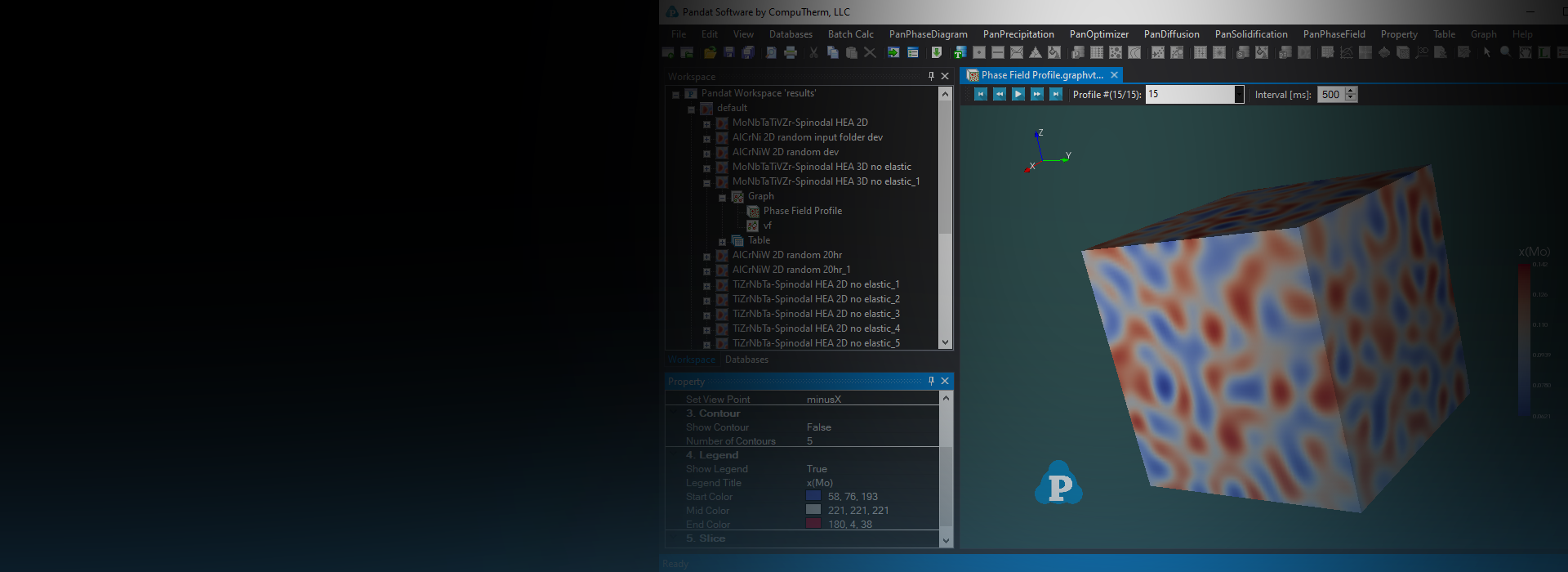
One Unified Workspace, Numerous Materials Data
Object-Oriented Design and Written in C++
Simple and Easy to Use API Functions
Flexible Materials Data Management
Customizable Table with Export Function
High Quality and Customizable Graph in 2D/3D
Pandat Software Modules
Pandat Software adopts modular design and enables users to easily switch between modules and perform various types of simulations in the same workspace.

- stable and metastable diagrams
- property contour diagrams
- thermodynamic properties
- chemical driving force
- material to material diagrams
- particle concurrent nucleation, growth and coarsening
- multi-phase co-precipitation
- precipitation and grain growth
- recrystallization and grain growth
- TTT and CCT curves
- diffusion couple
- homogenization
- carburization and decarburization
- particle dissolution
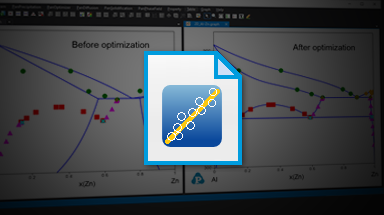
- Optimization of thermodynamic, kinetic and thermo-physical model parameters using experimental data
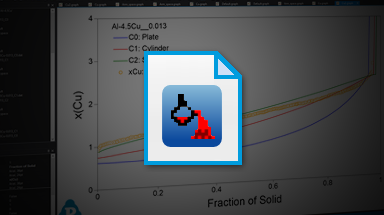
- solidification path
- back-diffusion in the solid
- dendrite arm coarsening
- micro-segregation
- solute trapping effect
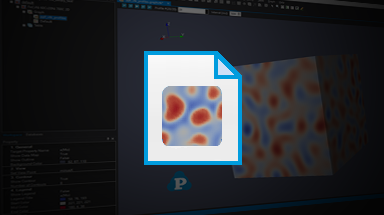
- direct coupling with CALPHAD
- feasible for multi-component alloys
- open architecture for user’s model plugin